Getting started
SNEEP is fast method to identify regulatory non-coding SNPs (rSNPs) that modify the binding sites of Transcription Factors (TFs) for large collections of SNPs provided by the user. SNEEP combines our statistical approach introduced in our preprint A statistical approach to identify regulatory DNA variants with epigenetic data and additional functionalities.
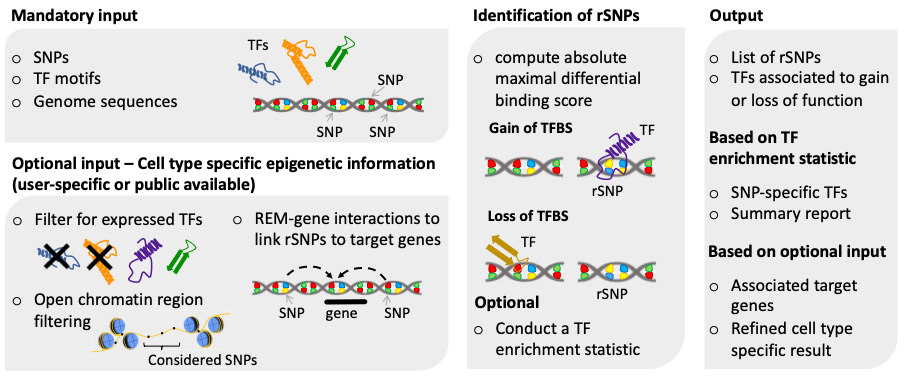
A graphical summary of SNEEP is shown below:
Installation
Bioconda package
We provide a bioconda package to install the main functionality of our approach. Therefore, an installation of Bioconda is needed.
conda install sneep
Manual installation
If you want to directly install SNEEP, please clone our GitHub repository and ensure that the following software is available on your machine:
c++11
g++ (9.3.0)
python3.x
bedtools (v2.27.1)
openmp
To build SNEEP, the following commands are used:
cd SNEEP/src/
make
Please add the path to our software (SNEEP/src) to our PATH environment (otherwise, internally called scrips might not be found).
We tested the code and the Makefile only on a Linux machine.
Testing your installation
We provide a test script to verify if your installation worked. To download the test data and scripts, please clone the latest version of our GitHub repository (if not already done)
git clone git@github.com:SchulzLab/SNEEP.git
download our Zenodo repository and unzip the files.
Additionally, a reference genome in FASTA format is needed. The different chromosomes within the file must be named as chr1, chr2. An example is shown below:
>chr1
ATCGGGTCA…
>chr2
TTTGAGACCAT…
For the provided examples in the following, please use genome version hg38.
To run our tests, please redirect into the SNEEP folder downloaded from GitHub and perform
bash runTests.sh <pathToGenome> <pathTodbSNP> <pathToInteractions>
where <pathTodbSNP> is the path to the dbSNPs_sorted.txt downloaded from our Zenodo repository and <pathToInteractions> needs to be the path to one of the interaction file, e.g., interactionsREM_PRO_HiC.txt
Basic usage
The following 4 files are required as minimal input to run SNEEP:
a file containing the TF motifs in TRANSFAC format,
a bedlike SNP file or a .VCF file,
a reference genome file in FASTA format,
a scale file providing motif-specific scale parameters to compute the significance of the effect of the SNP on the TF (provided).
Minimal example
To try SNEEP with the minimal required input, ensure that you are in the SNEEP folder and run:
differentialBindingAffinity_multipleSNPs examples/combined_Jaspar2022_Hocomoco_Kellis_human_transfac.txt examples/SNPs_EFO_0000612_myocardial_infarction.bed <path-to-genome-file> necessaryInputFiles/estimatedScalesPerMotif_1.9.txt
Per default, the result is stored in the directory ‘SNEEP_output’. The file ‘result.txt’ in the SNEEP output directory contains the predicted rSNPs. For more details about the resulting files, see the Section SNEEP result files. The run takes a couple of minutes using one core.
Detailed description of the required input files
We obtained human TF motifs from the JASPAR database (version 2022), HOCOMOCO and Kellis ENCODE database in the required format in the examples directory. Additionally, we provide the mouse motifs from the JASPAR database (version 2022) and the HOCOMOCO database. In general, our approach can be used for any species.
The required bedlike SNP file is a tab-separated file containing the following entries:
chr,
start position (0-based),
end position (0-based),
var1 (e.g., effector allele or alternative allele) ,
var2 (e.g., wildtype allele),
rsID if known; otherwise - ,
minor allele frequency (MAF) if known, otherwise -1. The minor allele frequency is important to provide if you want to assess the result of SNEEP against random controls. Then, SNEEP samples SNPs based on the MAF distribution of the input SNPs.
An example of a properly formated SNP file can be found below:
chr1 109274569 109274570 G A rs7528419 0.2009
chr1 109275907 109275908 C T rs646776 0.2384
chr1 154424939 154424940 G T rs12118721 1e-07
chr1 154424939 154424940 G T - 0.3
chr12 111569951 111569952 G C rs653178 -1
If you want to consider a SNP, that has multiple alternative alleles, such as for instance rs11206510 (T -> A,C,G) , please add one line per alternative allele to the bedlike SNP file. An example is shown below:
chr1 55030365 55030366 A T rs11206510 0.1018
chr1 55030365 55030366 C T rs11206510 0.1018
chr1 55030365 55030366 G T rs11206510 0.1018
Instead of the bedlike formated SNP file also a VCF can be given as input (ending with .vcf or .VCF). For more details of the file format, see here. We expect the first 8 columns to be given, where QUAL and FILTER can be set as “.”. If the rsID or the MAF is not known it is als set to “.”. An example is shown below:
##fileformat=VCFv4.0
#CHROM POS ID REF ALT QUAL FILTER INFO
1 154424940 rs12118721 T C,G . . MAF=1e-07
1 222638065 rs35700460 A G . . MAF=2e-11
1 222650187 rs17465637 A C . .
2 202881162 . T A . . .
An example file is provided within our GitHub repository (see examples/SNPs_EFO_0000612_myocardial_infarction.VCF).
SNEEP can only handle mutations affecting a single base pair (no deletions or insertions). Deletions and insertions are identified by the pipeline and ignored. Additionally, duplicated entries are only considered once.
In the reference genome file, the different chromosomes must be named as chr1, chr2, etc., resulting in the following format:
>chr1
ATCGGGTCA…
>chr2
TTTGAGACCAT…
For the provided examples in the following, please use genome version hg38. An already correctly formated file can, for instance, be downloaded from here filename hg38.fa.gz.
For human TF motifs, we provide a file containing motif-specific scale parameters. This information is necessary to determine whether a TF binding site is significantly affected by a SNP. Our modified Laplace distribution is dependent on two parameters, n, which is two times the length of the TF model, and the scale parameter b, which needs to be estimated. For the motif set we provide within our GitHub repository, we also estimated the scale parameter b listed in necessaryInputFiles/estimatedScalesPerMotif_1.9.txt. When a customized TF motif set is used, for instance, for a different species than humans, the scale parameter b needs to be estimated. Therefore, we provide a script XXX (TODO: provide more details here).